CommPath
CommPath is an R package for inference and analysis of ligand-receptor interactions from single cell RNA sequencing data.
Installation
CommPath R package can be easily installed from Github using devtools:
devtools::install_github("yingyonghui/CommPath")
library(CommPath)
Dependencies
Tutorials
In this vignette we show CommPath’s steps and functionalities for inference and analysis of ligand-receptor interactions by applying it to a scRNA-seq data (GEO accession number: GSE156337) on cells from hepatocellular carcinoma (HCC) patients.
Brief description of CommPath object
We start CommPath analysis by creating a CommPath object, which is a S4 object and consists of six slots including
(i) data, a matrix containing the normalized expression values by gene * cell;
(ii) cell.info, a data frame contain the information of cells;
(iii) meta.info, a list containing some important parameters used during the analysis;
(iv) LR.marker, a data.frame containing the result of differential expression test of ligands and receptors;
(v) interact, a list containing the information of LR interaction among clusters;
(vi) pathway, a list containing the information of pathways related to the ligands and receptors.
CommPath input
The expression matrix and cell indentity information are required for CommPath input. We downloaded the processed HCC scRNA-seq data from Mendeley data. For a fast review and illustration of CommPath’s functionalities, we randomly selected the expression data of 3000 cells across the top 5000 highly variable genes from the tumor and normal tissues, respectively. The example data are available in figshare.
We here illustrate the CommPath steps for date from the tumor tissues. And analysis for data from the normal tissues would be roughly in the same manner.
# load(url("https://figshare.com/ndownloader/files/33926126"))
load("path_to_download/HCC.tumor.3k.RData")
This dataset consists of 2 varibles which are required for CommPath input:
tumor.expr : expression matrix of gene * cell. Expression values are required to be first normalized by the library-size and log-transformed;
tumor.label : a vector of lables indicating identity classes of cells in the expression matrix, and the order of lables should match the order of cells in the expression matrix; usrs may also provide a data frame containing the meta infomation of cells with the row names matching the cells in the expression matrix and a column named as Cluster must be included to indicate identity classes of cells.
Identification of marker ligands and receptors
We start CommPath analysis by creating a CommPath object:
# Classify the species of the scRNA-seq experiment by the species parameter
# CommPath now enable the analysis of scRNA-seq experiment from human (hsapiens) and mouse (mmusculus).
tumor.obj <- createCommPath(expr.mat = tumor.expr,
cell.info = tumor.label,
species = 'hsapiens')
Firstly we’re supposed to identify marker ligands and receptors (ligands and receptors that are significantly highly expressed) for each identity class of cells in the expression matrix. CommPath provide findLRmarker to identify these markers by t.test or wilcox.test.
tumor.obj <- findLRmarker(object = tumor.obj, method = 'wilcox.test')
Identification of ligand-receptor (L-R) associations
# find significant L-R pairs
tumor.obj <- findLRpairs(object = tumor.obj,
logFC.thre = 0,
p.thre = 0.05)
The counts of significant LR pairs and overall interaction intensity among cell clusters are then stored in tumor.obj@interact[[‘InteractNumer’]],and the detailed information of each LR pair is stored in tumor.obj@interact[[‘InteractGeneUnfold’]].
Then you can visualize the interaction through a circos plot:
# Plot interaction for all cluster
circosPlot(object = tumor.obj)
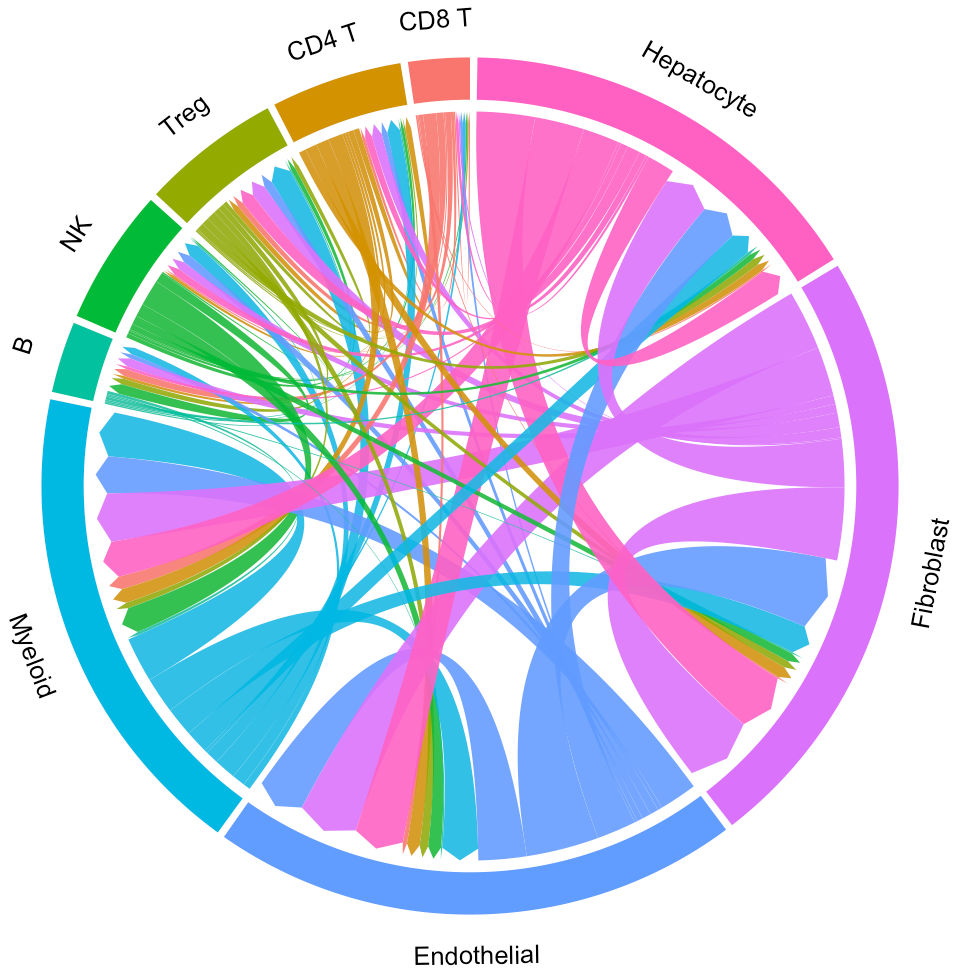
In the above circos plot, the directions of lines indicate the associations from ligands to receptors, and the widths of lines indicate the counts of LR pairs among clusters.
# Plot interaction for all cluster
circosPlot(object = tumor.obj)
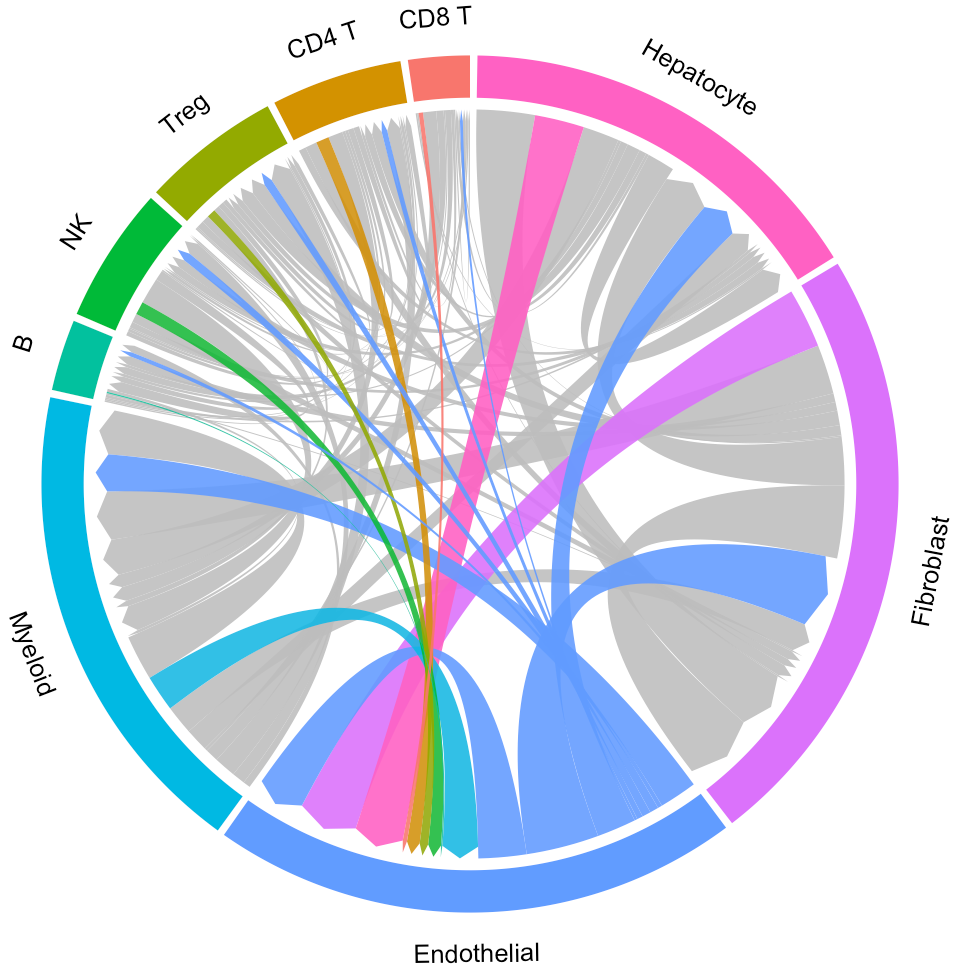
Now the widths of lines indicate the overall interaction intensity among clusters.
# Highlight the interaction of specific cluster
# Here we take the endothelial cell as an example
ident = 'Endothelial'
circosPlot(object = tumor.obj, ident = ident)
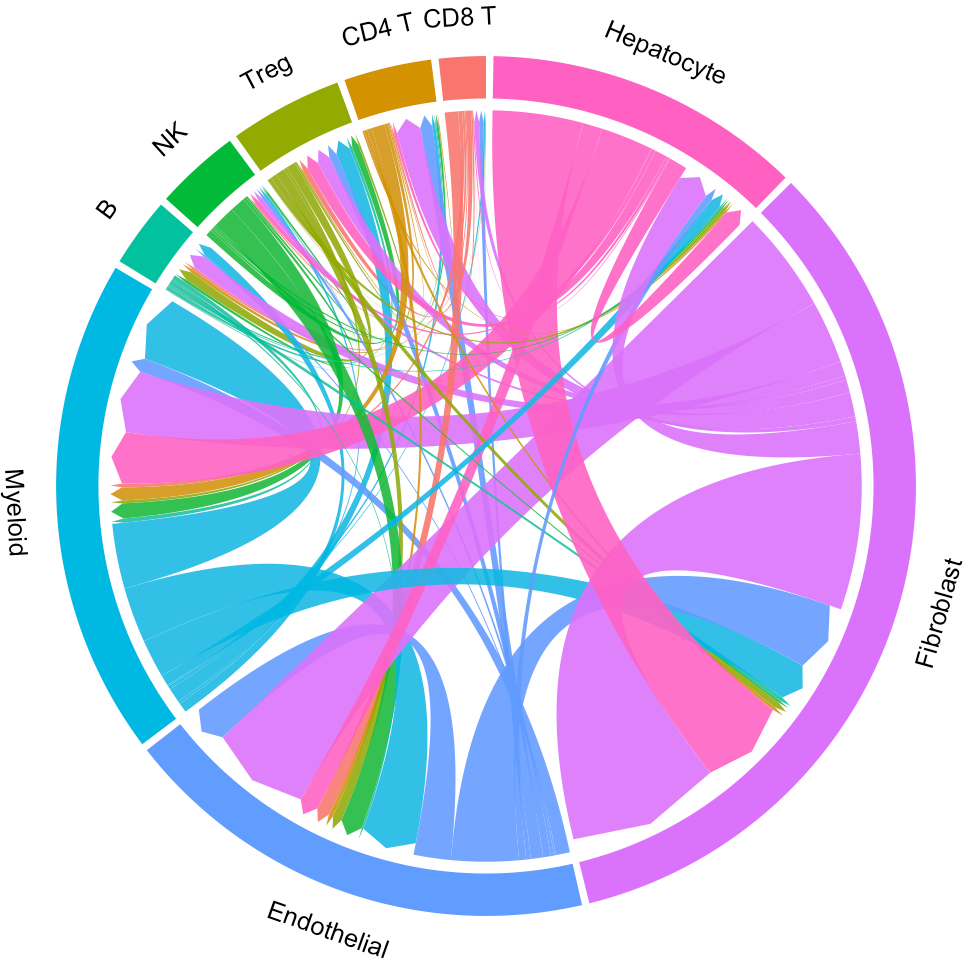
For a specific cluster of interest, CommPath provides function findLigand (findReceptor) to find the upstream (downstream) cluster and the corresponding ligand (receptor) for specific cluster and receptor (ligand):
# For the selected cluster and selected receptor, find the upstream cluster
select.ident = 'Endothelial'
select.receptor = 'ACKR1'
ident.up.dat <- findLigand(object = tumor.obj,
select.ident = select.ident,
select.receptor = select.receptor)
head(ident.up.dat)
# For the selected cluster and selected ligand, find the downstream cluster
select.ident = 'Endothelial'
select.ligand = 'CXCL12'
ident.down.dat <- findReceptor(object = tumor.obj,
select.ident = select.ident,
select.ligand = select.ligand)
head(ident.down.dat)
There are 7 columns stored in the variable ident.up.dat/ident.down.dat:
Columns Cell.From, Cell.To, Ligand, Receptor show the upstream and dowstream clusters and the specific ligands and receptors in the LR associations;
Columns Log2FC.LR, P.val.LR, P.val.adj.LR show the interaction intensity (measured by the product of Log2FCs of ligands and receptors)and the corresponding original and adjusted p value for hypothesis test of one pair of LR.
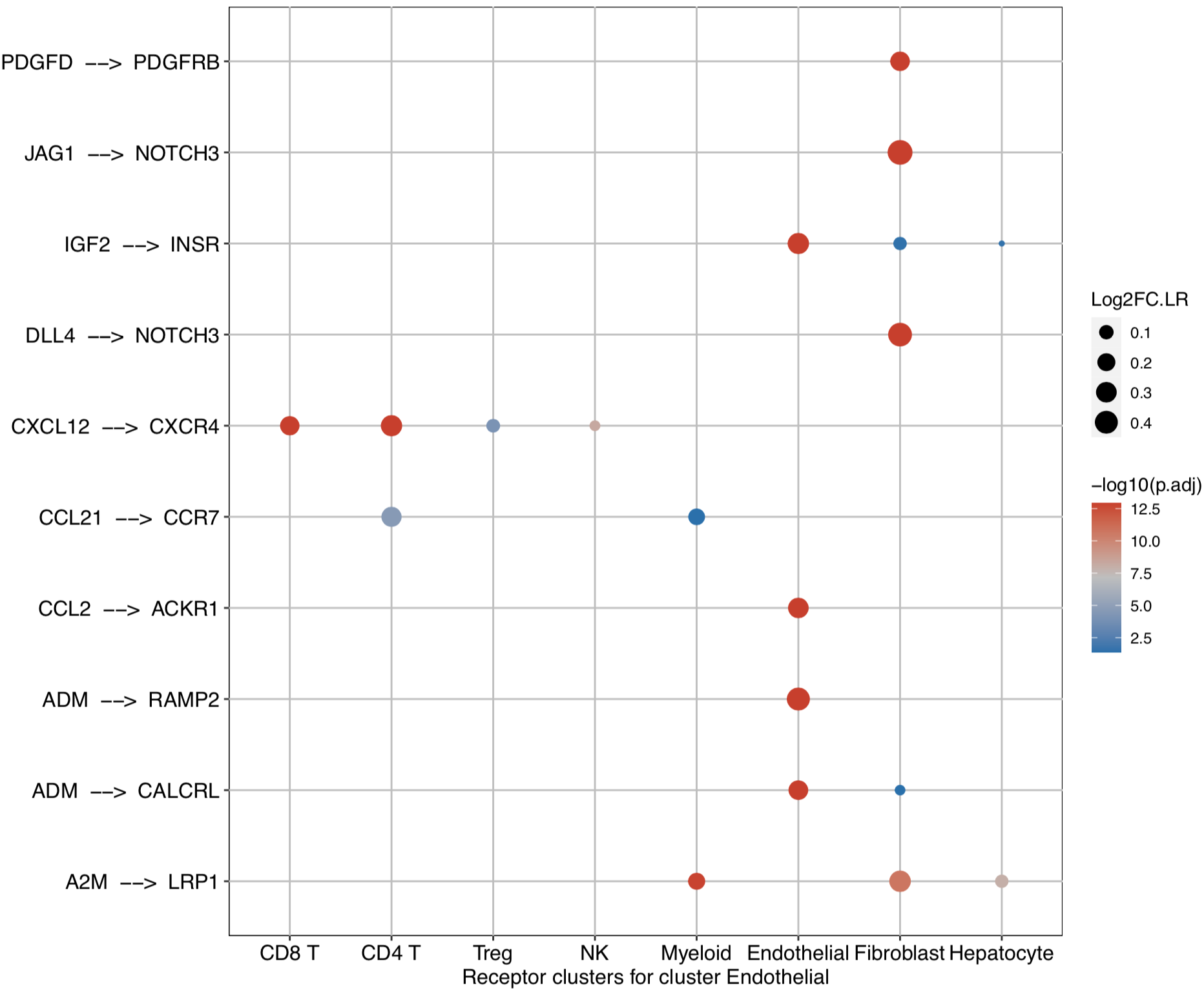
CommPath also provides dot plots to investigate its upstream clusters which release specific ligands and its downstream clusters which expressed specific receptors:
# Investigate the upstream clusters which release specific ligands to the interested cluster
dotPlot(object = tumor.obj, receptor.ident = ident)
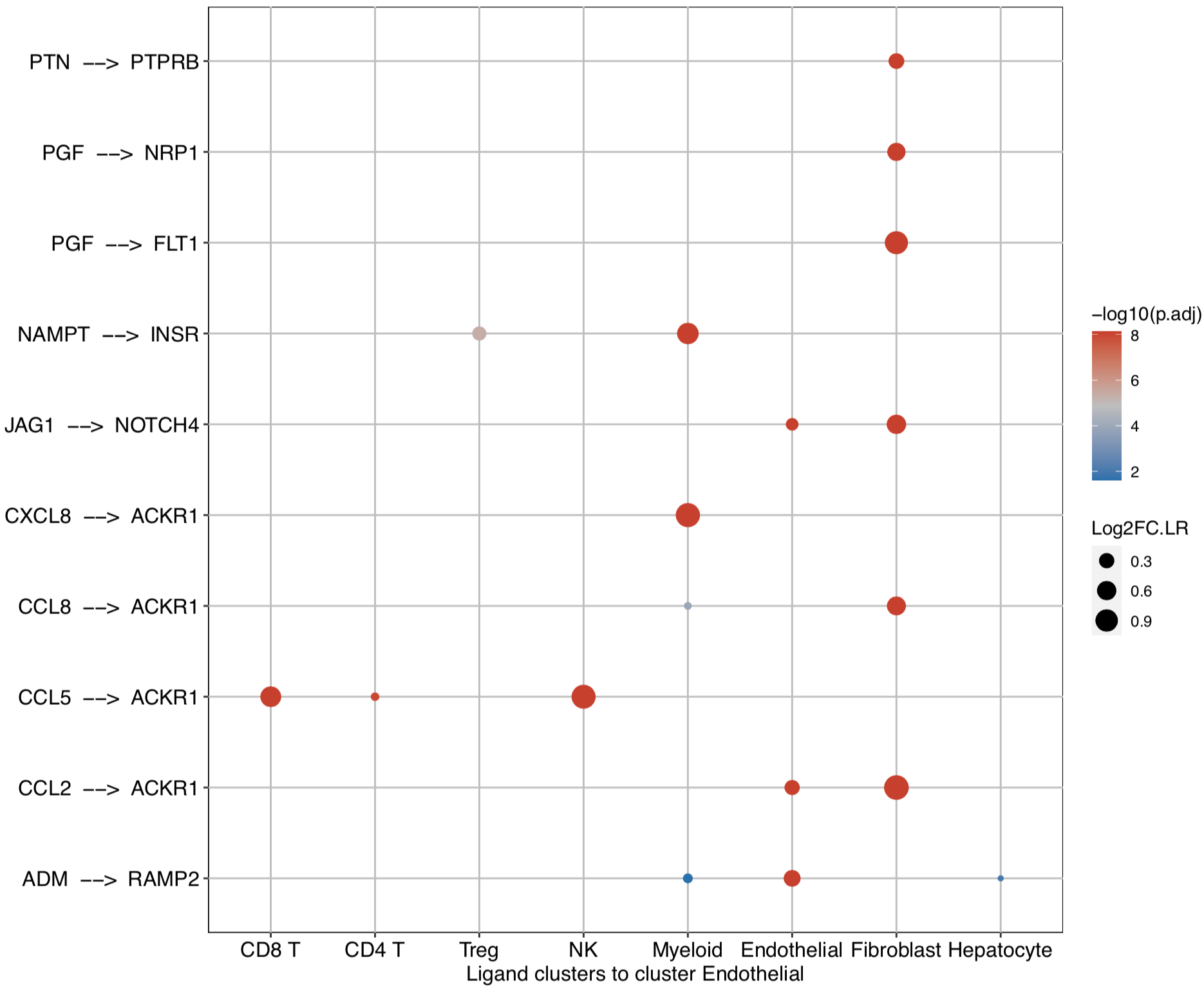
# Investigate the downstream clusters which expressed specific receptors for the interested cluster
dotPlot(object = tumor.obj, ligand.ident = ident)
Pathway analysis
CommPath conducts pathway analysis to identify signaling pathways involving the marker ligands and receptors for each cluster.
# Find pathways in which genesets show overlap with the marker ligands and receptors in the example dataset
# CommPath provides pathway annotations from KEGG pathways, WikiPathways, reactome pathways, and GO terms
tumor.obj <- findLRpath(object = tumor.obj, category = 'kegg')
Now genesets showing overlap with the marker ligands and receptors are stored in tumor.obj@interact[[‘pathwayLR’]]. Then we score the pathways to measure the activation levels for each pathway in each cell.
# Compute pathway activation score by the gsva algorithm or an average manner
# For more information about gsva algorithm, see the GSVA package
tumor.obj <- scorePath(object = tumor.obj, method = 'gsva', min.size = 10, parallel.sz = 4)
After that CommPath provide diffAllPath to perform pathway differential activation analysis for cells in each identity class and find the receptor and ligand in the pathway:
# get significantly up-regulated pathways in each identity class
acti.path.dat <- diffAllPath(object = tumor.obj, only.posi = TRUE, only.sig = TRUE)
head(acti.path.dat)
There are several columns stored in the variable acti.path.dat:
Columns mean.diff, mean.1, mean.2, t, df, p.val, p.val.adj show the statistic result; description shows the name of pathway;
Columns cell.up and ligand.up show the upstream identity classes which would release specific ligands to interact with the receptors from the current identity class;
Column receptor.in.path shows the marker receptors expressed by the current identity class and these receptors are included in the current pathway;
Column ligand.in.path shows the marker ligands released by the current identity class and these ligands are also included in the current pathway.
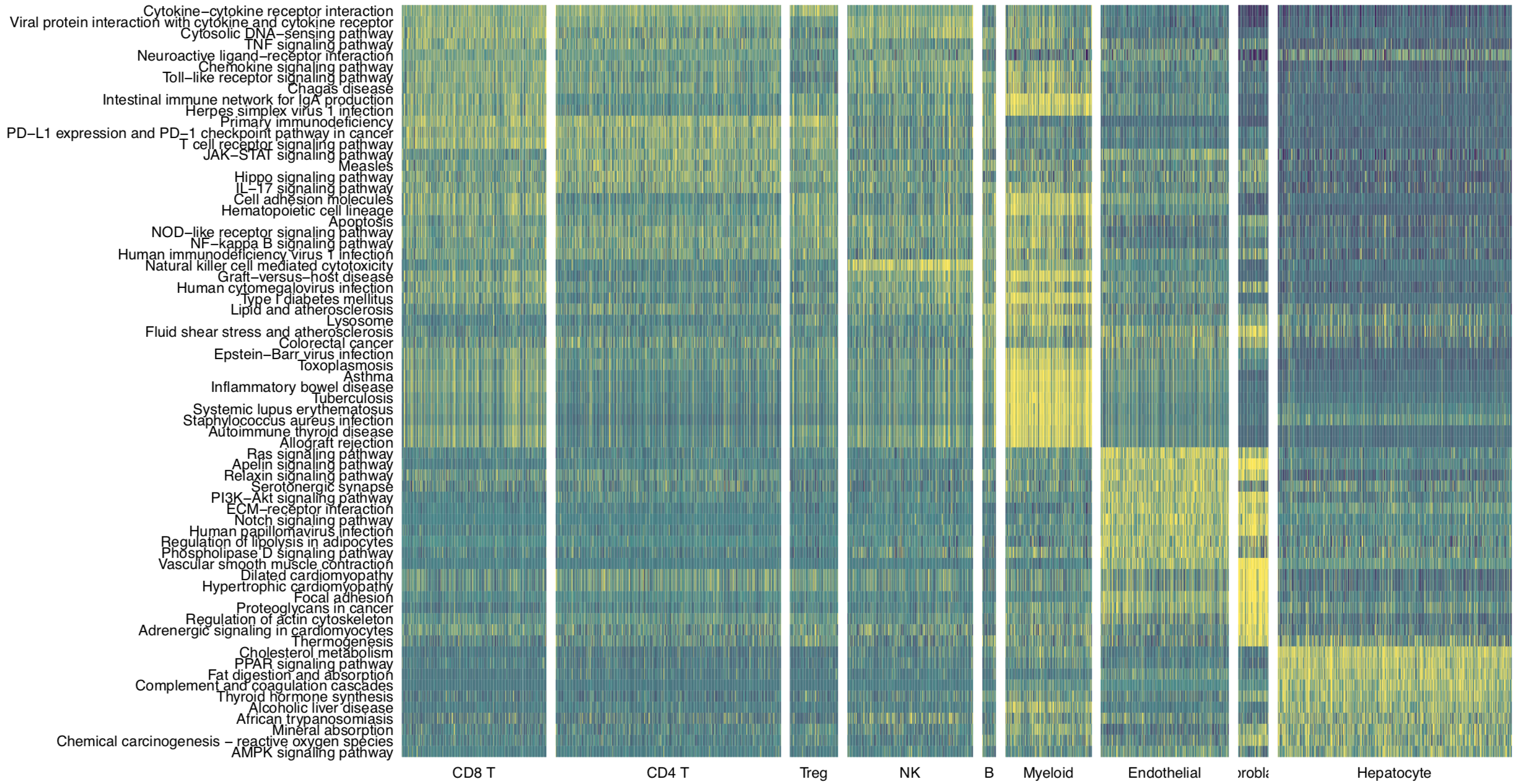
Then we use pathHeatmap to plot a heatmap of those differentially activated pathways for each cluster to display the highly variable pathways:
pathHeatmap(object = tumor.obj,
acti.path.dat = acti.path.dat,
top.n.pathway = 10,
sort = "p.val.adj")
Cell-cell interaction flow via pathways
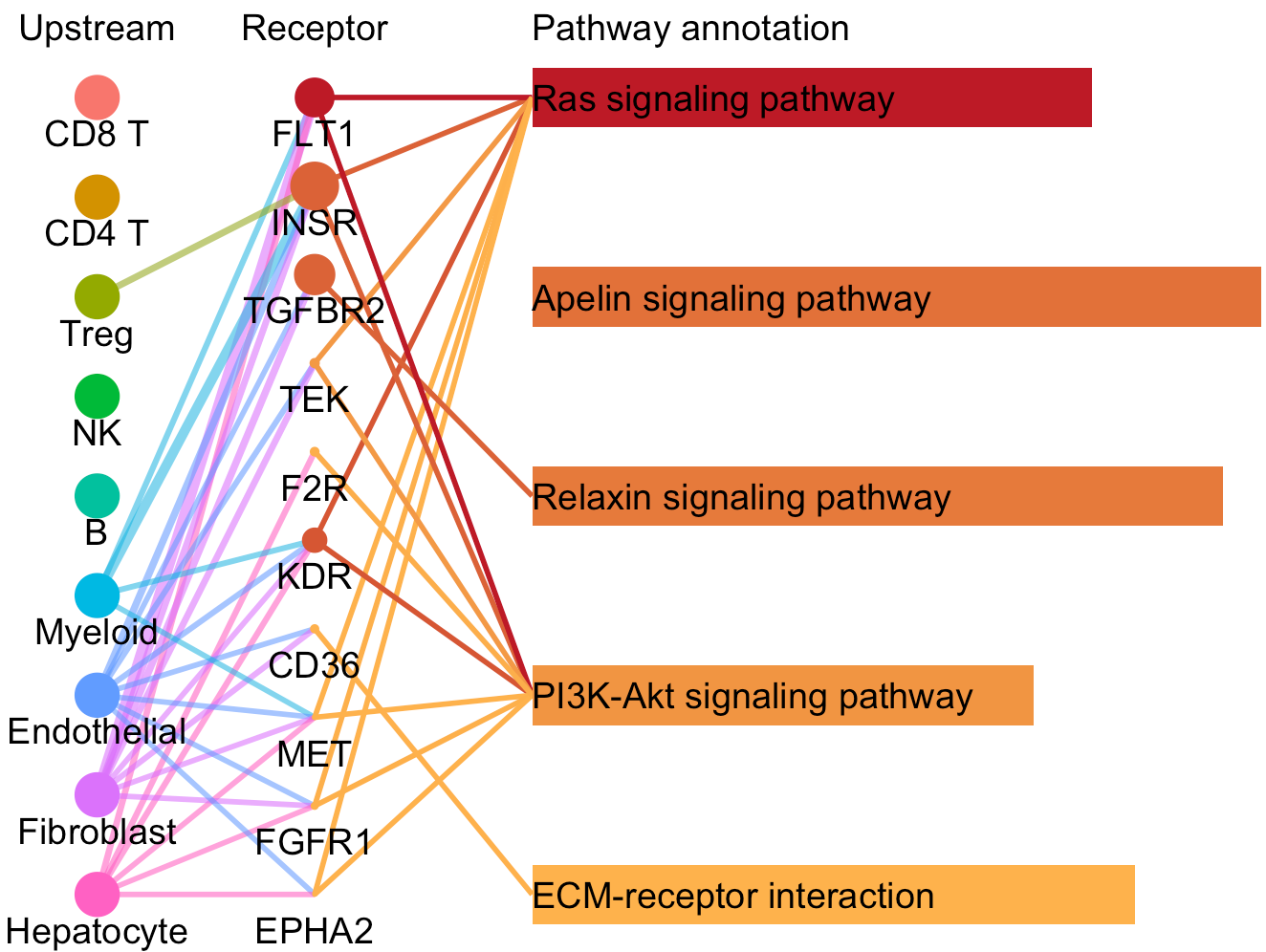
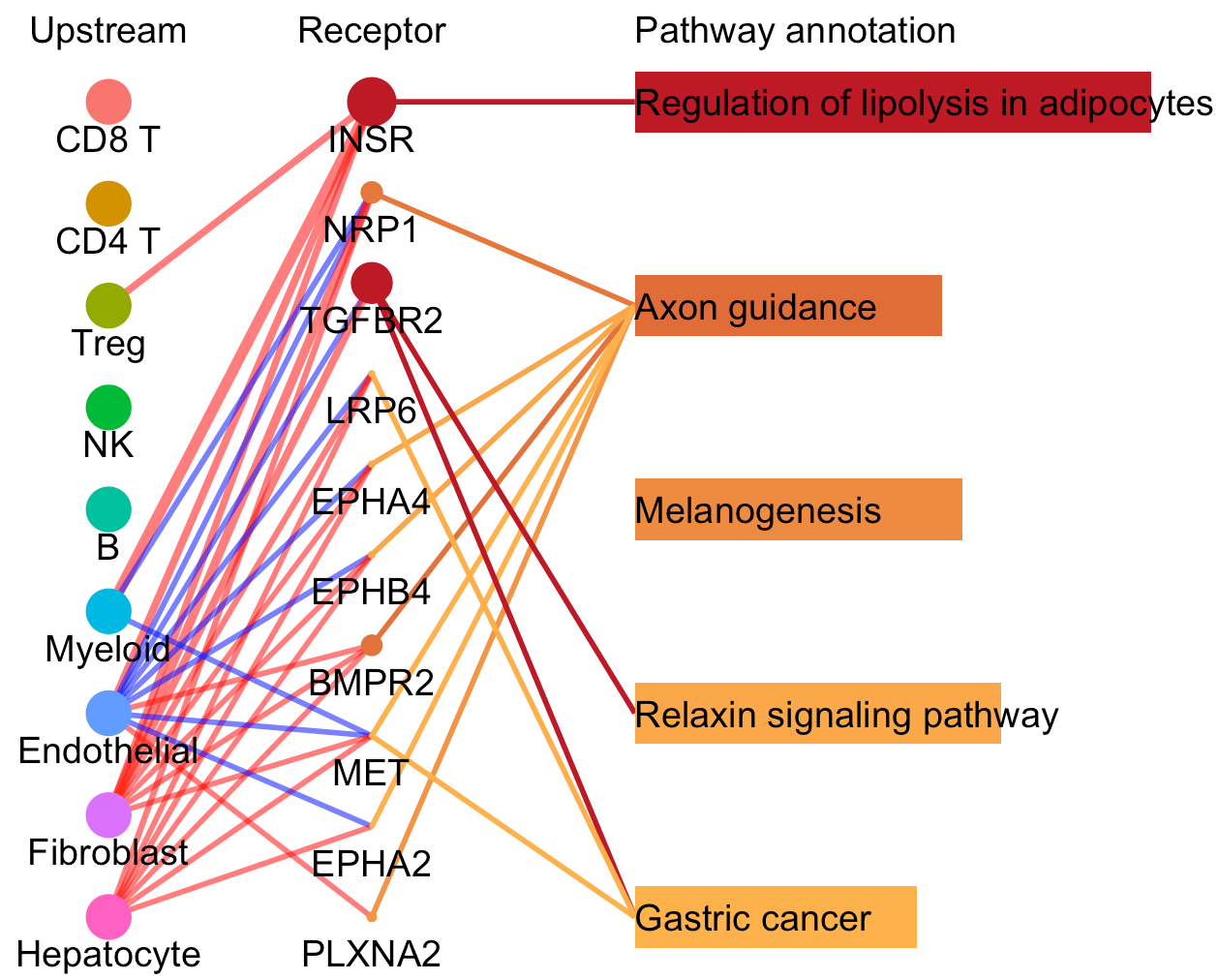
For a specific cell cluster, which here we name it as B for demonstration, CommPath identify the upstream cluster A sending signals to B, the downstream cluster C receiving signals from B, and the significantly activated pathways in B to mediate the A-B-C communication flow. More exactly, through LR and pathways analysis described above, CommPath is able to identify LR pairs between A and B, LR pairs between B and C, and pathways activated in B. Then CommPath screens for pathways in B which involve both the receptors to interact with A and ligands to interact with C.
# Identification and visualization of the identified pathways
# Plot to identify receptors and the associated activated pathways for a specific cluster
select.ident = 'Endothelial'
pathPlot(object = tumor.obj,
select.ident = select.ident,
acti.path.dat = acti.path.dat)
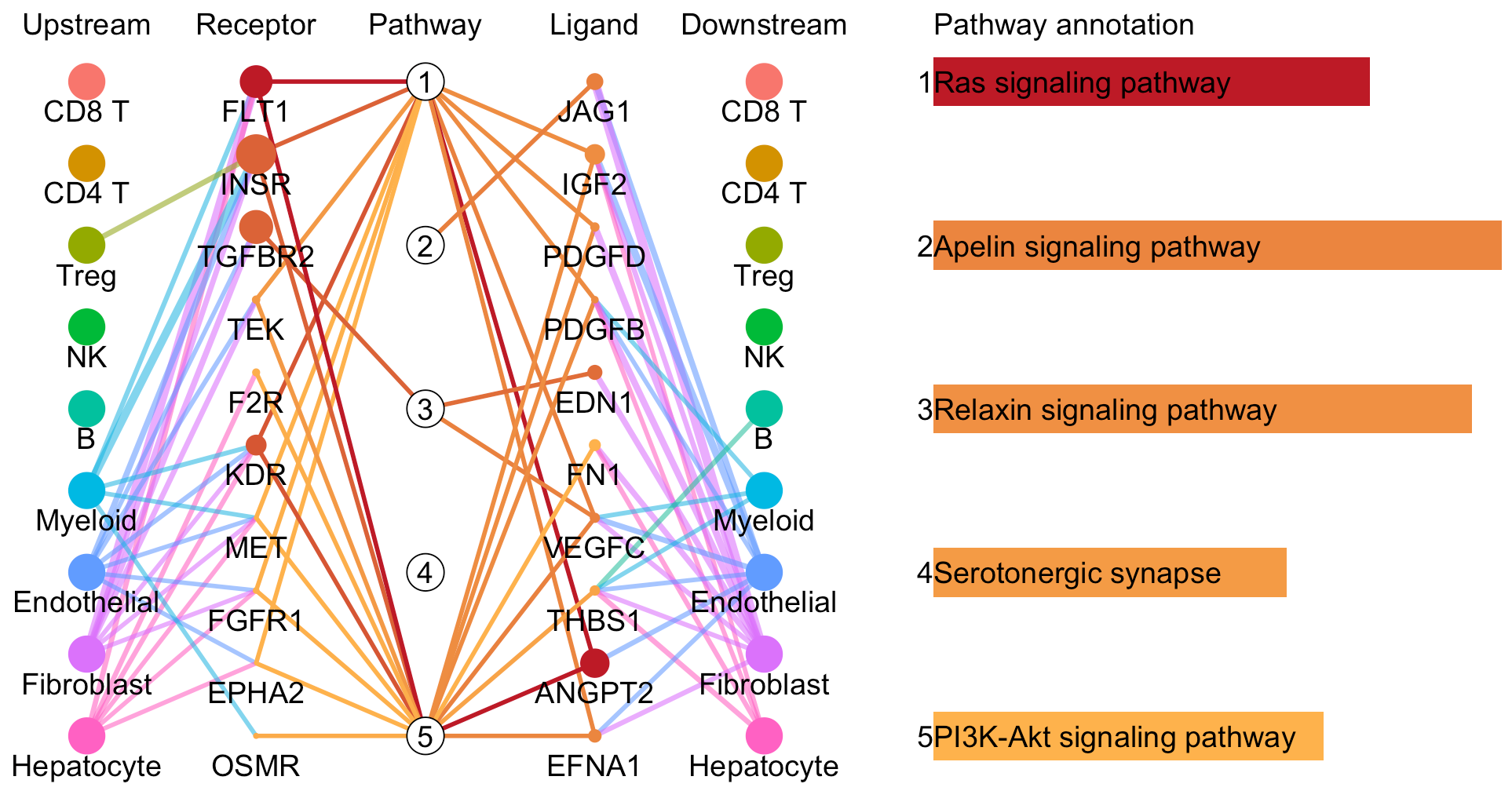
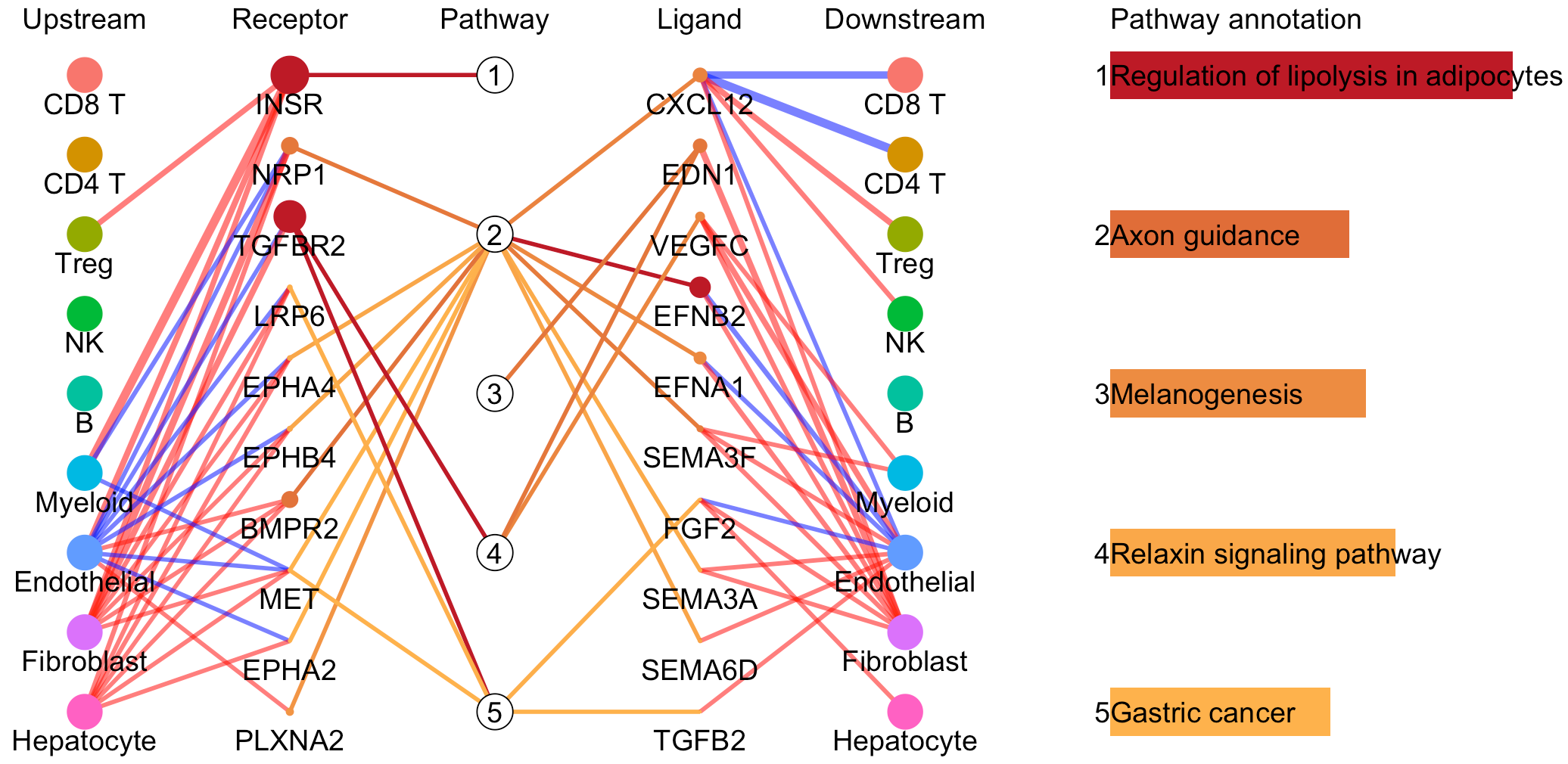
# Plot to identify receptors, the associated activated pathways, and the downstream clusters
pathInterPlot(object = tumor.obj,
select.ident = select.ident,
acti.path.dat = acti.path.dat)
Compare cell-cell interactions between two conditions
CommPath also provide useful utilities to compare cell-cell interactions between two conditions such as disease and control. Here we, for example, used CommPath to compare the cell-cell interactions between cells from HCC tumor and normal tissues. The example data from normal tissues are also available in figshare.
# load(url("https://figshare.com/ndownloader/files/33926129"))
load("path_to_download/HCC.normal.3k.RData")
We have pre-created the CommPath object for the normal samples following the above steps. This dataset consists of 3 varibles:
normal.expr : expression matrix for cells from normal tissues;
normal.label : indentity lables for cells from normal tissues;
normal.obj : CommPath object created from normal.expr and normal.label, and processed by CommPath steps described above.
To compare 2 CommPath object, we shall first identify the differentially expressed ligands and receptors, and differentially activated pathways between the same cluster of cells in the two object.
# Take endothelial cells as example
# Identification of differentially expressed ligands and receptors
diff.marker.dat <- diffCommPathMarker(object.1 = tumor.obj, object.2 = normal.obj, select.ident = 'Endothelial')
# Identification of differentially activated pathways
diff.path.dat <- diffCommPathPath(object.1 = tumor.obj, object.2 = normal.obj, select.ident = 'Endothelial', parallel.sz = 4)
Then we compare the differentially activated pathways and the cell-cell communication flow mediated by those pathways.
# To compare differentially activated pathways and the involved receptors between the selected clusters of two CommPath object
pathPlot.compare(object.1 = tumor.obj, object.2 = normal.obj, select.ident = 'Endothelial', diff.marker.dat = diff.marker.dat, diff.path.dat = diff.path.dat)
# To compare the pathway mediated cell-cell communication flow for a specific cluster between 2 CommPath object
pathInterPlot.compare(object.1 = tumor.obj, object.2 = normal.obj, select.ident = 'Endothelial', diff.marker.dat = diff.marker.dat, diff.path.dat = diff.path.dat)
sessionInfo()
R version 4.0.3 (2020-10-10)
Platform: x86_64-conda-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS/LAPACK: /home/luh/miniconda3/envs/seurat4/lib/libopenblasp-r0.3.17.so
locale:
[1] LC_CTYPE=zh_CN.UTF-8 LC_NUMERIC=C
[3] LC_TIME=zh_CN.UTF-8 LC_COLLATE=zh_CN.UTF-8
[5] LC_MONETARY=zh_CN.UTF-8 LC_MESSAGES=zh_CN.UTF-8
[7] LC_PAPER=zh_CN.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=zh_CN.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] GSVA_1.38.2 ggplot2_3.3.5 dplyr_1.0.7 reshape2_1.4.4
[5] circlize_0.4.13 CommPath_0.1.0
loaded via a namespace (and not attached):
[1] Rcpp_1.0.7 lattice_0.20-44
[3] digest_0.6.27 assertthat_0.2.1
[5] utf8_1.2.2 R6_2.5.1
[7] GenomeInfoDb_1.26.7 plyr_1.8.6
[9] stats4_4.0.3 RSQLite_2.2.8
[11] httr_1.4.2 pillar_1.6.2
[13] zlibbioc_1.36.0 GlobalOptions_0.1.2
[15] rlang_0.4.11 annotate_1.68.0
[17] blob_1.2.2 S4Vectors_0.28.1
[19] Matrix_1.3-4 labeling_0.4.2
[21] BiocParallel_1.24.1 stringr_1.4.0
[23] RCurl_1.98-1.4 bit_4.0.4
[25] munsell_0.5.0 DelayedArray_0.16.3
[27] compiler_4.0.3 pkgconfig_2.0.3
[29] BiocGenerics_0.36.1 shape_1.4.6
[31] tidyselect_1.1.1 SummarizedExperiment_1.20.0
[33] tibble_3.1.3 GenomeInfoDbData_1.2.4
[35] IRanges_2.24.1 matrixStats_0.60.1
[37] XML_3.99-0.7 fansi_0.5.0
[39] crayon_1.4.1 withr_2.4.2
[41] bitops_1.0-7 grid_4.0.3
[43] xtable_1.8-4 GSEABase_1.52.1
[45] gtable_0.3.0 lifecycle_1.0.0
[47] DBI_1.1.1 magrittr_2.0.1
[49] scales_1.1.1 graph_1.68.0
[51] stringi_1.7.4 cachem_1.0.6
[53] farver_2.1.0 XVector_0.30.0
[55] ellipsis_0.3.2 generics_0.1.0
[57] vctrs_0.3.8 tools_4.0.3
[59] bit64_4.0.5 Biobase_2.50.0
[61] glue_1.4.2 purrr_0.3.4
[63] MatrixGenerics_1.2.1 parallel_4.0.3
[65] fastmap_1.1.0 AnnotationDbi_1.52.0
[67] colorspace_2.0-2 GenomicRanges_1.42.0
[69] memoise_2.0.0
Is this can be used for snRNA-Seq data analysis of plant
Yes, you just need prepare a a matrix containing the normalized expression values by gene * cell